

 English
English Bahasa Melayu
Bahasa Melayu Bahasa Indonesia
Bahasa Indonesia Tiếng Việt
Tiếng Việt ไทย
ไทย
本翻譯僅作學術交流用,無商業意圖,請勿轉載,如有疑議問請來信
本研究發現,透過每日僅攝取600千卡的嚴格飲食計劃,第2型糖尿病患者的胰島素β細胞功能得以正常化。該方法顯著減少了患者胰腺和肝臟的三酸甘油酯含量,改善了胰島素抵抗問題。這項研究挑戰了傳統觀點,證明透過飲食控制,第2型糖尿病的病情可以實現逆轉。

第2型糖尿病的逆轉:與減少胰腺和肝臟三酸甘油酯相關的胰島素β細胞功能正常化
Reversal of type 2 diabetes: normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol
Lim EL, Hollingsworth KG, Aribisala BS, Chen MJ, Mathers JC, Taylor R. Reversal of type 2 diabetes: normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia. 2011 Oct;54(10):2506-14. doi: 10.1007/s00125-011-2204-7. Epub 2011 Jun 9. PMID: 21656330; PMCID: PMC3168743.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3168743/
Abstract
Aims/hypothesis
Type 2 diabetes is regarded as inevitably progressive, with irreversible beta cell failure. The hypothesis was tested that both beta cell failure and insulin resistance can be reversed by dietary restriction of energy intake.
Methods
Eleven people with type 2 diabetes (49.5 ± 2.5 years, BMI 33.6 ± 1.2 kg/m2, nine male and two female) were studied before and after 1, 4 and 8 weeks of a 2.5 MJ (600 kcal)/day diet. Basal hepatic glucose output, hepatic and peripheral insulin sensitivity and beta cell function were measured. Pancreas and liver triacylglycerol content was measured using three-point Dixon magnetic resonance imaging. An age-, sex- and weight-matched group of eight non-diabetic participants was studied.
Results
After 1 week of restricted energy intake, fasting plasma glucose normalised in the diabetic group (from 9.2 ± 0.4 to 5.9 ± 0.4 mmol/l; p = 0.003). Insulin suppression of hepatic glucose output improved from 43 ± 4% to 74 ± 5% (p = 0.003 vs baseline; controls 68 ± 5%). Hepatic triacylglycerol content fell from 12.8 ± 2.4% in the diabetic group to 2.9 ± 0.2% by week 8 (p = 0.003). The first-phase insulin response increased during the study period (0.19 ± 0.02 to 0.46 ± 0.07 nmol min−1 m−2; p < 0.001) and approached control values (0.62 ± 0.15 nmol min−1 m−2; p = 0.42). Maximal insulin response became supranormal at 8 weeks (1.37 ± 0.27 vs controls 1.15 ± 0.18 nmol min−1 m−2). Pancreatic triacylglycerol decreased from 8.0 ± 1.6% to 6.2 ± 1.1% (p = 0.03).
Conclusions/interpretation
Normalisation of both beta cell function and hepatic insulin sensitivity in type 2 diabetes was achieved by dietary energy restriction alone. This was associated with decreased pancreatic and liver triacylglycerol stores. The abnormalities underlying type 2 diabetes are reversible by reducing dietary energy intake.
Keywords
Insulin secretion, Liver fat, Low energy diet, Pancreatic fat, Type 2 diabetes
摘要
目的/假設
第2型糖尿病被認為是不可逆轉的,具有不可逆的胰島素β細胞功能衰竭。本研究測試了透過限制能量攝入的飲食,可以逆轉β細胞功能衰竭和胰島素抵抗的假設。
方法
研究了11名第2型糖尿病患者(年齡49.5 ± 2.5歲,BMI 33.6 ± 1.2 kg/m²,九男二女),在開始2.5 MJ(600千卡)/天飲食前後的第1、4和8週進行了研究。測量了基礎肝葡萄糖輸出、肝臟和周邊胰島素敏感性以及β細胞功能。使用三點Dixon磁共振成像測量了胰腺和肝臟的三酸甘油酯含量。進行了與年齡、性別和體重相匹配的8名非糖尿病參與者的研究。
結果
限制能量攝入1週後,糖尿病組的空腹血漿葡萄糖正常化(從9.2 ± 0.4 mmol/l降至5.9 ± 0.4 mmol/l;p = 0.003)。胰島素對肝葡萄糖輸出的抑制從43 ± 4%提高至74 ± 5%(與基線相比p = 0.003;對照組為68 ± 5%)。糖尿病組的肝臟三酸甘油酯含量從12.8 ± 2.4%降至第8週的2.9 ± 0.2%(p = 0.003)。研究期間,第一階段胰島素反應增加(從0.19 ± 0.02增至0.46 ± 0.07 nmol min−1 m−2;p < 0.001)並接近對照值(0.62 ± 0.15 nmol min−1 m−2;p = 0.42)。第8週,最大胰島素反應變得超常規(1.37 ± 0.27對照組為1.15 ± 0.18 nmol min−1 m−2)。胰腺三酸甘油酯從8.0 ± 1.6%降至6.2 ± 1.1%(p = 0.03)。
結論/解釋
僅通過限制飲食能量達到了第2型糖尿病的胰島素β細胞功能和肝臟胰島素敏感性的正常化。這與減少的胰腺和肝臟三酸甘油酯儲存有關。第2型糖尿病的基本異常可以通過減少飲食能量攝入來逆轉。
關鍵詞
胰島素分泌,肝脂肪,低能量飲食,胰腺脂肪,第2型糖尿病
引言
長期以來,第2型糖尿病被認為是一種慢性進展性疾病,能夠緩解但無法治愈。血漿葡萄糖濃度無論控制程度或治療類型如何都會持續上升[1]。β細胞功能隨時間線性下降,10年後超過50%的個體需要胰島素治療[2]。β細胞功能的變化已被充分描述[3, 4],而在第2型糖尿病過程中β細胞質量穩定減少[5, 6]。總體來看,有強有力的證據表明第2型糖尿病是不可逆轉的進展性疾病,最終很可能需要胰島素治療以維持良好的血糖控制。
然而,第2型糖尿病在接受胃旁路手術後明顯可逆[7]。血漿葡萄糖濃度在手術後幾天內正常化,遠在顯著體重減輕之前,人們普遍認為胃腸手術的保護作用是通過改變分泌激素的分泌所介導[8, 9]。其他研究者已證明通過中度能量限制改善第2型糖尿病的血糖控制[10]。我們假設突然的負能量平衡對代謝的深刻影響可以解釋胃旁路手術後的效果[11],具體來說,肝臟細胞內脂肪酸濃度的降低會導致向胰腺輸出的脂蛋白三酸甘油酯減少,從而使β細胞擺脫了長期過量脂肪酸暴露的抑制效應。
本研究旨在驗證僅靠急性負能量平衡可逆轉第2型糖尿病,通過正常化β細胞功能和胰島素敏感性的假說。我們檢驗了第一階段和總胰島素反應的恢復以及肝臟和周邊胰島素敏感性。此外,為了探討觀察到的結果的機制基礎,我們量化了胰腺和肝臟脂肪含量的變化。
方法
參與者
招募了第2型糖尿病患者(年齡35-65歲,HbA1c 6.5-9.0% [48-75毫摩爾/摩爾],糖尿病病程<4年,穩定的BMI 25-45 kg/m²)。被排除在外的參與者包括正在接受噻唑烷二酮類藥物、胰島素、類固醇或β阻滯劑治療的人,血清肌酐>150毫摩爾/升,血清谷丙轉氨酶水平超過參考範圍上限的2.5倍,或有MRI禁忌症的人。他們持續接受他汀類藥物治療。該研究方案已獲得紐卡斯爾泰恩河畔倫理委員會第2號的批准,所有參與者均已知情同意。硫脲尿病藥物(2名參與者)在基線研究前2個月停用,甲福明(7名參與者)在基線研究前1週停用。飲食遵從性使用毛細血管酮體水平(Xceed Optium;艾伯特糖尿病護理,英國梅登黑德)進行評估。三名參與者未能遵守飲食(兩名在第一週期間,一名在第4-8週期間),一名因與研究無關的醫療原因退出研究。因此,共有11名參與者(9男2女,年齡49.5 ± 2.5歲)完成了研究。
還研究了9名與體重、年齡和性別相匹配的對照參與者(7男2女,年齡49.7 ± 2.5歲;表1)。這些參與者沒有糖尿病家族史,沒有服用任何藥物,並且通過標準75克口服葡萄糖耐量試驗(OGTT)確認具有正常的葡萄糖代謝。
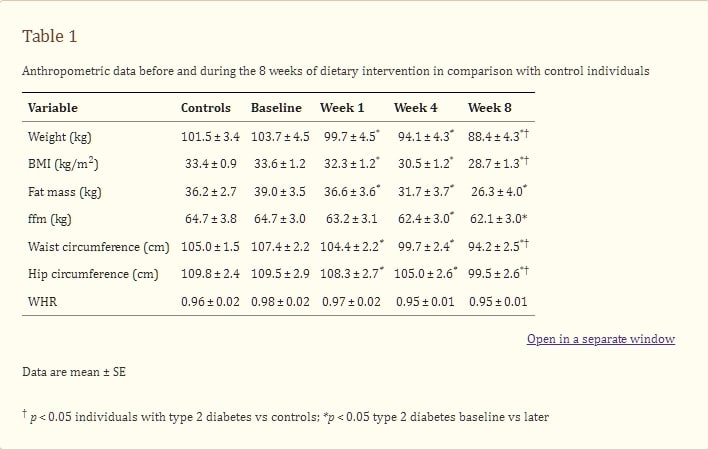
表1飲食干預8週前及期間的人體測量數據與對照組個體的比較
實驗方案
參與者被要求在研究開始前繼續他們慣常的飲食模式。在飲食干預前(第-1天)進行基線測量,並在非常低能量飲食後的第1、4和8週對β細胞功能、胰島素敏感性、肝臟和胰腺脂肪含量以及總體脂肪進行評估。一組配對的非糖尿病對照組僅在一個時間點進行研究,無飲食干預。
基線測量後,第2型糖尿病患者開始飲食,該飲食包括液體飲食配方(46.4%碳水化合物,32.5%蛋白質和20.1%脂肪;維生素、礦物質和微量元素;每天2.1 MJ(510千卡);Optifast;雀巢營養,英國克羅伊登)。這被補充了三份非澱粉類蔬菜,使總能量攝入量約為每天2.5 MJ(600千卡)。參與者被提供蔬菜食譜建議,通過變化日常飲食來提高遵從性。他們還被鼓勵每天至少喝2升水或其他無能量飲料,並要求保持他們慣常的體育活動水平。通過定期電話聯繫提供持續的支持和鼓勵。在8週干預結束後,參與者恢復正常飲食,但提供了有關份量大小和健康飲食的信息。
第8週出現的顯著結果要求進行實驗性的後續跟進,並獲得了額外的倫理許可,以在完成飲食干預後12週重複進行MRI研究和進行OGTT。
肝臟葡萄糖產生和胰島素敏感性
經過一夜禁食後,將導管插入肘前靜脈進行輸液,並在對側腕靜脈採集動脈化血樣。使用[6′6′-2H]葡萄糖(98%富集;劍橋同位素實驗室,安多佛,馬薩諸塞州,美國)來確定肝臟葡萄糖產生[12, 13],並在150分鐘基礎期的最後30分鐘計算基礎率。整個過程中同位素的預輸注富集量不顯著。在0分鐘時開始等血糖-高胰島素鉗夾(胰島素輸注率為40 mU m−2 min−1)[14]。使用等血糖是為了確保在每個研究時間點都能觀察到每位參與者的真實禁食狀態。每位參與者在基礎期結束時觀察到的血糖水平下進行鉗夾。在高胰島素葡萄糖鉗夾的最後30分鐘確定全身胰島素敏感性,作為校正了葡萄糖空間和尿液損失的全身葡萄糖處置[14, 15]。在穩態條件下計算葡萄糖代謝清除率,方法是將全身胰島素敏感性除以穩態血漿葡萄糖。
β細胞功能評估
鉗夾試驗後60分鐘,通過初始葡萄糖劑量和可變20%葡萄糖輸注[16]實現了兩個連續的30分鐘方波步驟高血糖(比基線高2.8和5.6毫摩爾/升)。每2分鐘採集一次血樣,用於測定血漿葡萄糖、胰島素和C肽濃度,為期10分鐘,其後每5分鐘採集一次,為期其他20分鐘。在第二階段高血糖期間給予精氨酸脈衝劑量,隨後每2分鐘採樣10分鐘。使用計算機程序實施去卷積的正則化方法[17],並使用C肽動力學的群體模型[18],計算胰島素分泌率。
磁共振測量
使用飛利浦3.0 T Achieva掃描儀和六通道心臟線圈(飛利浦醫療保健,荷蘭貝斯特)進行三個梯度回波掃描,具有相鄰的失相和同相回波(重複時間/回波時間/平均次數/翻轉角度=50毫秒/3.45、4.60、5.75毫秒/1/30°,矩陣160×109,視野範圍400-480毫米以適應參與者尺寸,相位視野70%)。在17秒屏氣內獲得六個切片,以覆蓋肝臟(切片厚度10毫米)和胰腺(切片厚度5毫米)。數據在MATLAB中(MathWorks,英國劍橋)進行分析,以產生單獨的脂肪和水圖像[19]。圖像的脂肪含量表示為每個體素總信號的百分比。一名觀察員(K. G. Hollingsworth)以盲測方式定義並平均評估五個肝臟感興趣區域和兩個胰腺感興趣區域的器官內脂肪百分比。胰腺脂肪數據僅代表胰腺內脂肪,肝臟數據避免了血管和膽囊的污染。這是通過圖像獲取後的數據處理實現的,允許從解剖切片中手動選擇完全器官內部體積。掃描間的Bland-Altman重複性係數為肝臟0.5%和胰腺0.9%。
體組成和人體測量
使用空氣位移容積描記法(BOD POD Express;Life Measurement,康科德,加州,美國)在經過一夜禁食後測量體脂百分比。在參與者放鬆站立姿勢下測量腰圍和臀圍。腰圍測量點為前上髖骨棘和肋骨下緣中點,臀圍測量點為股骨大轉子水平。整個研究期間,所有測量均由單一觀察員(E. L. Lim)進行。
分析程序
血漿葡萄糖濃度使用葡萄糖氧化酶法測量(YSI葡萄糖分析儀;Yellow Springs,俄亥俄州,美國),血漿胰島素和C肽濃度使用ELISA法測量(Dako,Ely,英國),血漿三酸甘油酯使用脂酶法測量,釋放的甘油由羅氏Cobas離心分析儀使用比色測定法測量(ABX Diagnostics,蒙彼利埃,法國),HbA1c由Biorad HPLC測量(TOSOH Corporation,東京,日本)。血漿葡萄糖中的2H原子百分比過量使用Thermo ‘Voyager’單四極質譜儀和Thermo ‘Trace’氣相色譜儀測定(Thermo Scientific,沃爾瑟姆,馬薩諸塞州,美國)。
統計方法
使用SPSS 17.0軟件(SPSS Inc.,芝加哥,伊利諾伊州,美國)進行統計分析。數據以平均值±標準誤表示。使用學生t檢驗進行糖尿病和對照組之間的統計比較,而組內差異使用配對t檢驗確定。實驗中連續數據的變化通過重複測量單向方差分析評估,並在適當時進行事後Bonferroni檢驗。使用Spearman等級檢驗檢查相關性。統計顯著性接受標準為p<0.05。
結果
血漿葡萄糖和胰島素
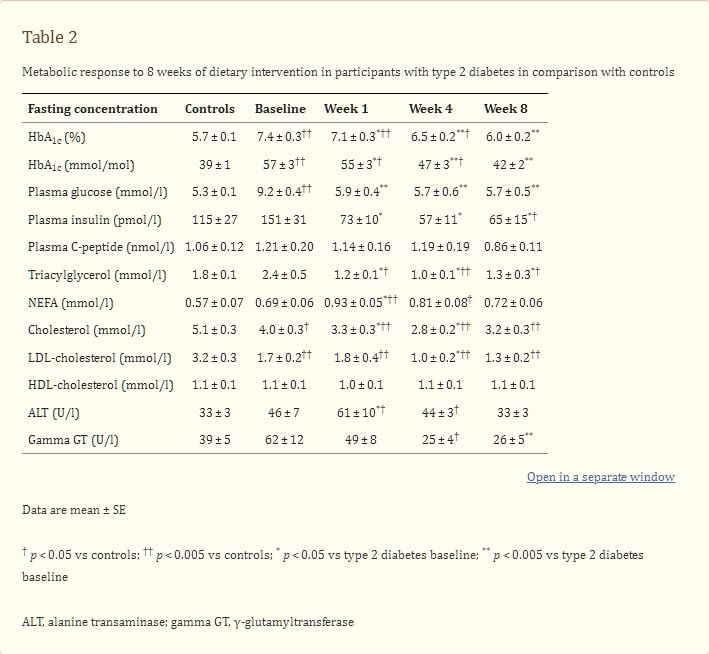
飲食干預1週後,空腹血漿葡萄糖從9.2 ± 0.4毫摩爾/升降至5.9 ± 0.4毫摩爾/升(p = 0.003;圖1),與非糖尿病對照組(5.3 ± 0.1毫摩爾/升)無顯著差異(p = 0.18)。在剩餘的8週研究期間保持穩定(第4週和第8週為5.7 ± 0.5毫摩爾/升;與對照組相比p = 0.52)。HbA1c從7.4 ± 0.3%(57 ± 3毫摩爾/摩爾)降低,在第8週與非糖尿病對照值無顯著差異(6.0 ± 0.2%對5.7 ± 0.1% [42 ± 2對39 ± 1毫摩爾/摩爾],p = 0.27)。空腹血漿胰島素從151 ± 31降至73 ± 10皮摩爾/升(p = 0.03),到第8週降至65 ± 15皮摩爾/升(與基線相比p = 0.03;與對照組相比p = 0.04)。空腹血漿C肽也有類似下降(表2)。
圖1{kind=link}
飲食干預8週對(a)血漿葡萄糖,(b)肝葡萄糖產生(HGP)和(c)肝三酸甘油酯含量(TG)的影響,用於糖尿病參與者(黑色三角形)。白色圓圈表示體重匹配的非糖尿病對照組的平均值。數據以平均值±標準誤顯示。
在等血糖鉗夾期間,對照組的血漿葡萄糖被鉗定在4.6 ± 0.1毫摩爾/升,而糖尿病組在基線時為7.0 ± 0.3毫摩爾/升(p < 0.001)。在第1週,糖尿病個體達到的鉗定血漿葡萄糖水平為4.8 ± 0.2毫摩爾/升(與基線相比p < 0.001),第4週和第8週時類似(分別為4.8 ± 0.3和4.8 ± 0.4,與基線相比p < 0.001)。
肝胰島素敏感性和肝三酸甘油酯含量
能量限制的第一週,基礎肝葡萄糖產生顯著減少(從2.40 ± 0.28減少到1.59 ± 0.07 mg kgffm−1 min−1;p = 0.05),與基線相比在研究剩餘時間內持續減少,在第8週與對照組相比無顯著差異(分別為1.71 ± 0.11與2.11 ± 0.22 mg kgffm−1 min−1;p = 0.60;圖1)。在基線時,糖尿病組的肝胰島素敏感性,通過胰島素輸注抑制肝葡萄糖產生評估,為43 ± 4%,與對照組的68 ± 5%相比(p = 0.001)。在能量限制的第一週,肝胰島素反應性顯著改善,胰島素對肝葡萄糖產生的抑制增加到74 ± 5%(與基線相比p = 0.003)。
干預第1週期間,肝三酸甘油酯含量下降了30 ± 5%(p < 0.001),變得與對照值相似(p = 0.75)。在干預期間持續下降,達到非肥胖個體的正常範圍[20](2.9 ± 0.2%;p = 0.003;圖1),即總減少70 ± 5%。在基線時,對照組的肝三酸甘油酯含量為8.5 ± 1.9%,與糖尿病組的12.8 ± 2.4%相比(p = 0.14)。在對照組中,基線肝三酸甘油酯與BMI相關(Rs = 0.71;p < 0.05),但在糖尿病組中則不相關(Rs = −0.50;p = 0.12)。
β細胞對葡萄糖的敏感性和胰腺三酸甘油酯含量
空腹胰島素分泌率在第一週從0.10 ± 0.01降至0.06 ± 0.01 nmol min−1 m−2(p < 0.05),此後保持穩定。在胰島素分泌測試期間,計劃的血漿葡萄糖濃度增加+2.8和+5.6毫摩爾/升被實現(圖2)。在糖尿病個體中,6分鐘時的胰島素分泌峰值在基線時最小(0.19 ± 0.02對照0.62 ± 0.15 nmol min−1 m−2;p < 0.001;圖2)。第一階段胰島素反應穩定增加,在第8週與基線相比顯著不同(第1、4和8週分別為0.29 ± 0.05、0.34 ± 0.06和0.46 ± 0.07 nmol min−1 m−2;p = 0.20、p = 0.09、p = 0.006)。在第8週,第2型糖尿病個體的胰島素分泌率與對照組無顯著差異(0.46 ± 0.07對0.62 ± 0.15 nmol min−1 m−2;p = 0.42)。第1週後精氨酸誘導的胰島素反應增加(從0.72 ± 0.11增至0.95 ± 0.19 nmol min−1 m−2;p < 0.006),到第8週完全正常化(1.37 ± 0.27對1.15 ± 0.18 nmol min−1 m−2;與對照組相比p = 0.77;與基線相比p < 0.03;圖2)。在基線時,糖尿病參與者的反應比對照組低38%(0.72 ± 0.11對1.15 ± 0.18 nmol min−1 m−2;p = 0.04)。
圖2{kind=link}
對照組和糖尿病參與者在每個時間點的胰島素分泌測試數據。a 每組達到的血漿葡萄糖水平。在(b)非糖尿病對照組,(c)糖尿病組基線,(d)糖尿病組飲食第1週,(e)糖尿病組第4週和(f)糖尿病組第8週獲得的胰島素分泌率(ISR)。數據以平均值±標準誤顯示。
糖尿病組的胰腺三酸甘油酯含量為8.0 ± 1.6%,在8週後穩定下降至6.2 ± 1.1%(p = 0.03;圖3)。在對照個體中,胰腺三酸甘油酯含量為6.0 ± 1.3%(與第2型糖尿病參與者在基線時相比p = 0.17)。在對照組和糖尿病組中,胰腺三酸甘油酯含量與BMI無相關性(分別為Rs = 0.31, p = 0.36;Rs = 0.01, p = 0.98)。
圖3{kind=link}
a 糖尿病個體在8週飲食干預期間第一階段胰島素反應的變化,以及(b)胰腺三酸甘油酯(TG)含量的變化(黑色三角形)。白色圓圈表示體重匹配的非糖尿病對照組的平均值。數據以平均值±標準誤顯示。
周邊胰島素敏感性
整個研究期間,周邊胰島素敏感性以葡萄糖處置率表示並無顯著變化。胰島素刺激的葡萄糖處置率在基線和第8週分別為3.83 ± 0.23和4.36 ± 0.36 mg kgffm−1 min−1(其中ffm代表無脂肪質量)(p = 0.21)。為了校正研究天數之間鉗夾葡萄糖水平的差異,檢查了葡萄糖代謝清除率的變化。基線與第1週之間空腹血漿葡萄糖下降。飲食干預在第1週或第4週對葡萄糖代謝清除率均無顯著影響(分別為3.1 ± 0.3與4.23 ± 0.34和4.21 ± 0.36 ml kgffm−1 min−1),但到第8週時可見改善(5.2 ± 0.5 ml kgffm−1 min−1;基線與第8週相比p = 0.003;對照組為5.2 ± 0.4 ml kgffm−1 min−1;p = 0.98)。
體重和體組成
8週飲食干預期間平均體重減輕15.3 ± 1.2公斤,相當於初始體重的15 ± 1%(表1)。第一週減重3.9 ± 0.2公斤(其中61%為脂肪減少),第1至4週間減重5.7 ± 0.6公斤(86%為脂肪),最後4週減重5.7 ± 0.7公斤(94%為脂肪)。腰圍和臀圍同等程度減少,8週期間腰臀比(WHR)保持不變(表1)。
血漿脂質
飲食能量限制的第一週,血漿三酸甘油酯水平降低了一半(從2.4 ± 0.5降至1.2 ± 0.1毫摩爾/升;p < 0.02),此後保持穩定(表2)。總膽固醇也降低,而高密度脂蛋白膽固醇在研究期間保持不變(表2)。空腹血漿非酯化脂肪酸(NEFA)水平在糖尿病參與者中略高於基線時與之配對的對照組(0.69 ± 0.06對0.57 ± 0.07毫摩爾/升;p = 0.24),但差異不顯著。在研究期間,糖尿病參與者的空腹NEFA在第1週顯著增加(0.93 ± 0.05毫摩爾/升;與基線相比p = 0.03)。隨著持續的低能量攝入,血漿NEFA穩步下降至接近基線值(第4週和第8週分別為0.81 ± 0.08和0.72 ± 0.06毫摩爾/升)。
干預後觀察
在完成飲食干預後12週的隨訪中,平均體重增加了3.1 ± 1.0公斤。肝三酸甘油酯保持低水平且不變(2.9 ± 0.2對3.0 ± 0.3%;p = 0.80),胰腺三酸甘油酯進一步小幅下降(6.2 ± 1.1對5.7 ± 1.1%;p = 0.005)。HbA1c保持不變(6.0 ± 0.2對6.2 ± 0.1% [42 ± 2對44 ± 1毫摩爾/摩爾];p = 0.10),空腹血漿葡萄糖略微增加(5.7 ± 0.5對6.1 ± 0.2毫摩爾/升;p < 0.01),75克口服葡萄糖耐量試驗(OGTT)2小時血漿葡萄糖為10.3 ± 1.0毫摩爾/升。有三名參與者出現糖尿病復發,這是根據OGTT後2小時血漿葡萄糖>11.1毫摩爾/升判斷的。空腹血漿胰島素濃度保持不變(57 ± 11對65 ± 15皮摩爾/升),空腹血漿NEFA進一步降低(0.72 ± 0.06對0.54 ± 0.05毫摩爾/升;p < 0.02)。一位參與者因接受卵巢囊腫(非惡性)手術而未能參加重新測試。
討論
本研究證明,第2型糖尿病背後的β細胞功能衰竭和胰島素抵抗這兩個雙重缺陷可以僅通過急性負能量平衡來逆轉。觀察到一種反應層次,肝臟胰島素敏感性的改變非常早,而β細胞功能的改變較慢。在減少能量攝入的最初7天內,空腹血糖和肝臟胰島素敏感性降至正常,肝內脂肪減少了30%。在8週的飲食能量限制期間,β細胞功能增加接近正常,胰腺脂肪減少。干預後,參與者在12週內體重增加了3.1 ± 1.0公斤,但他們的HbA1c保持穩定,而胰腺和肝臟的脂肪含量沒有增加。這些數據與假設一致,即胰島素分泌和胰島素抵抗的異常,這些是第2型糖尿病的根本原因,有一個共同的病因,即肝臟和胰腺的過量脂肪累積[11]。這提供了一個統一的假設來解釋此前似乎需要影響胰腺和胰島素敏感組織的單獨疾病過程的常見疾病。
對血漿葡萄糖上升反應缺乏快速胰島素分泌是第2型糖尿病的標誌[3, 21],而β細胞功能的下降決定了對胰島素治療的需求進展[2]。然而,即使使用磺脲尿病藥,傳統治療也未能產生超過小幅增加第一階段胰島素反應。因此,本研究中對飲食能量限制的胰島素反應的迅速和程度的恢復令人矚目。它支持了關於脂肪酸對體外和體內胰島素分泌抑制作用的累積信息[22-24],並且是人類中第一個直接證據,證明第2型糖尿病的β細胞缺陷可以通過持續的負能量平衡逆轉。人類長期高濃度血漿脂肪酸會降低胰島素分泌[25, 26],並且先前已經顯示胰腺脂肪含量與第2型糖尿病有關[27-29]。在啮齒動物自發性糖尿病發病前,整個胰腺的總脂肪和胰島三酸甘油酯含量都急劇增加[30, 31]。在體外,長期飽和脂肪酸暴露會抑制β細胞對葡萄糖的急性胰島素反應,去除脂肪酸後可以恢復這種反應[32]。
目前的數據提供了明確的證據,表明降低整體胰腺脂肪與β細胞功能的恢復相關。然而,對β細胞功能的負面影響可能是由毒性中間體如二酰甘油和神經酰胺施加的,這些物質會對急性代謝變化迅速反應[33],而不是由儲存的三酸甘油酯本身,後者作為脂肪酸中間體濃度的指標。胰島素分泌指標與胰腺脂肪之間沒有相關性,這表明對這些毒性中間體的耐受度有個體閾值,而不是胰腺內簡單的劑量-反應關係。
空腹血漿葡萄糖濃度由肝葡萄糖產生率決定,肝胰島素敏感性與肝內脂肪含量成反比[13, 34–36]。先前的研究已顯示適度減重與肝內脂肪含量的下降相關[10, 37]。Petersen等人先前報告稱,經過8週適度能量限制的第2型糖尿病患者,在等血糖高胰島素鉗夾測量中,肝臟胰島素敏感性改善,而肌肉胰島素敏感性無顯著變化[10]。在健康肥胖個體中,非常低的能量攝入已觀察到在幾天內降低肝脂肪含量[38]。本研究首次展示了在第2型糖尿病中,飲食限制對肝脂肪儲存和肝葡萄糖產生的早期時間進程下降。周邊胰島素敏感性的變化在早期血糖正常化中沒有發揮作用。低熱量飲食第1週後血漿NEFA的急劇升高可能阻止了周邊胰島素敏感性的變化,儘管這並未阻止肝臟胰島素敏感性的迅速改善。
我們先前提出了一種假說,即建立伴隨第2型糖尿病逆轉的病理生理變化將揭示決定該病發病序列的變化[11]。這種雙循環假說是根據胃旁路手術後的觀察得出的,特別是證明了胰胃分流術後空腹血糖濃度在幾天內下降的事實[39]。由於這些變化在顯著體重減輕之前發生,人們普遍接受了胃旁路手術通過改變分泌素激素產生如此迅速的效果。然而,分泌素的變化程度是適度的,在第2型糖尿病中並非總是存在,且在胃帶束縛術後缺失[40-43]。對胃旁路手術後突然負能量平衡對葡萄糖代謝的潛在作用關注甚少,儘管最近一項小型研究表明,手術後第一週胰島素抵抗的改善可單獨歸因於負能量平衡[44]。已報告胃旁路手術後與低熱量飲食相比分泌素樣肽-1分泌增加,但在存在胃腸吻合術的情況下使用的口服葡萄糖負荷可能解釋了血漿葡萄糖和分泌素的早期迅速上升,偶爾伴有早期倒流症狀[45]。
必須考慮本研究的限制。首先,樣本量必須足夠小,以便應用金標準代謝研究方法和磁共振技術進行檢查。然而,參與者來自第2型糖尿病人群,他們的臨床和人體測量特徵對於這種病狀是典型的。
其次,胰腺脂肪測量包括器官內脂肪細胞的脂肪含量,因為目前的方法無法評估更具機械意義的胰島細胞內脂肪酸含量。動物數據表明這兩個變量是相關的[31]。儘管糖尿病組的胰腺脂肪含量高出30%,但本研究的目的是展示對飲食干預的反應,而不是測試與體重匹配的非糖尿病個體的差異。在本研究中檢查的BMI限制範圍內,沒有觀察到胰腺脂肪與BMI之間的相關性。
第三,參與者被選擇為患有相對較短時間的第2型糖尿病(最多4年),進一步研究必須確定病程較長的第2型糖尿病可逆性的程度。此外,還需要進行進一步研究,以確定在恢復正常飲食後12週觀察到的血糖調節的長期結果,因為這些觀察必然是有限的。
最後,儘管一些對照組參與者在代謝正常的情況下肝脂肪水平升高,可以從最近對PNPLA3基因[46]的描述中考慮。該基因的G等位基因決定了高肝脂肪水平,但這種形式與代謝異常無關。這為觀察到的個體易感性差異提供了明確的遺傳基礎,儘管肝脂肪含量增加,也不易於胰島素抵抗,並部分解釋了第2型糖尿病和對照組之間重疊的肝脂肪水平。可能還有其他尚未界定的遺傳因素影響個體對胰腺脂肪累積的易感性差異,尤其是在抑制葡萄糖依賴性胰島素分泌方面。
本研究首次展示了第2型糖尿病患者通過急性限制飲食能量攝入恢復正常β細胞功能和肝葡萄糖輸出的時間進程。這些變化與胰腺和肝三酸甘油酯濃度的降低相關。這種新的洞見使我們能夠理解個體和人群中第2型糖尿病的因果關係。這對於給剛診斷的患者提供的信息具有重大意義,他們應該知道自己患有一種潛在可逆的狀況,而不是不可避免地進展的狀況。
參考文獻
1. UK Prospective Diabetes Study Group Intensive blood glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) Lancet. 1999;352:837–853. [PubMed] [Google Scholar]
2. Prospective Diabetes Study Group UK. Overview of 6 years’ therapy of type II diabetes: a progressive disease. UK Prospective Diabetes Study 16. Diabetes. 1995;44:1249–1258. doi: 10.2337/diabetes.44.11.1249. [PubMed] [CrossRef] [Google Scholar]
3. Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest. 1999;104:787–794. doi: 10.1172/JCI7231. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
4. Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia. 2003;46:3–19. doi: 10.1007/s00125-003-1190-9. [PubMed] [CrossRef] [Google Scholar]
5. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [PubMed] [CrossRef] [Google Scholar]
6. Hanley SC, Austin E, Assouline-Thomas B, et al. {beta}-Cell mass dynamics and islet cell plasticity in human type 2 diabetes. Endocrinology. 2010;151:1462–1472. doi: 10.1210/en.2009-1277. [PubMed] [CrossRef] [Google Scholar]
7. Pories WJ, Caro JF, Flickinger EG, Meelheim HD, Swanson MS. The control of diabetes mellitus (NIDDM) in the morbidly obese with the Greenville Gastric Bypass. Ann Surg. 1987;206:316–323. doi: 10.1097/00000658-198709000-00009. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
8. Rubino F, Forgione A, Cummings DE, et al. The mechanism of diabetes control after gastrointestinal bypass surgery reveals a role of the proximal small intestine in the pathophysiology of type 2 diabetes. Ann Surg. 2006;244:741–749. doi: 10.1097/01.sla.0000224726.61448.1b. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
9. Kashyap SR, Daud S, Kelly KR, et al. Acute effects of gastric bypass versus gastric restrictive surgery on beta-cell function and insulinotropic hormones in severely obese patients with type 2 diabetes. Int J Obes (Lond) 2010;34:462–471. doi: 10.1038/ijo.2009.254. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
10. Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes. 2005;54:603–608. doi: 10.2337/diabetes.54.3.603. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
11. Taylor R. Pathogenesis of type 2 diabetes: tracing the reverse route from cure to cause. Diabetologia. 2008;51:1781–1789. doi: 10.1007/s00125-008-1116-7. [PubMed] [CrossRef] [Google Scholar]
12. Hother-Nielsen O, Henriksen JE, Holst JJ, Beck-Nielsen H. Effects of insulin on glucose turnover rates in vivo: isotope dilution versus constant specific activity technique. Metabolism. 1996;45:82–91. doi: 10.1016/S0026-0495(96)90204-8. [PubMed] [CrossRef] [Google Scholar]
13. Ravikumar B, Gerrard J, Dalla Man C, et al. Pioglitazone decreases fasting and postprandial endogenous glucose production in proportion to decrease in hepatic triglyceride content. Diabetes. 2008;57:2288–2295. doi: 10.2337/db07-1828. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
14. DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237:E214–E223. [PubMed] [Google Scholar]
15. Rizza RA, Mandarino LJ, Gerich JE. Dose–response characteristics for effects of insulin on production and utilization of glucose in man. Am J Physiol. 1981;240:E630–E639. [PubMed] [Google Scholar]
16. Toschi E, Camastra S, Sironi AM, et al. Effect of acute hyperglycemia on insulin secretion in humans. Diabetes. 2002;51(Suppl 1):S130–S133. doi: 10.2337/diabetes.51.2007.S130. [PubMed] [CrossRef] [Google Scholar]
17. Hovorka R, Soons PA, Young MA. ISEC: a program to calculate insulin secretion. Comput Meth Programs Biomed. 1996;50:253–264. doi: 10.1016/0169-2607(96)01755-5. [PubMed] [CrossRef] [Google Scholar]
18. Van Cauter E, Mestrez F, Sturis J, Polonsky KS. Estimation of insulin secretion rates from C-peptide levels. Comparison of individual and standard kinetic parameters for C-peptide clearance. Diabetes. 1992;41:368–377. doi: 10.2337/diabetes.41.3.368. [PubMed] [CrossRef] [Google Scholar]
19. Glover GH, Schneider E. Three-point Dixon technique for true water/fat decomposition with B0 inhomogeneity correction. Magn Reson Med. 1991;18:371–383. doi: 10.1002/mrm.1910180211. [PubMed] [CrossRef] [Google Scholar]
20. Szczepaniak LS, Nurenberg P, Leonard D, et al. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab. 2005;288:E462–E468. doi: 10.1152/ajpendo.00064.2004. [PubMed] [CrossRef] [Google Scholar]
21. Pfeifer MA, Halter JB, Porte D. Insulin secretion in diabetes mellitus. Am J Med. 1981;70:579–588. doi: 10.1016/0002-9343(81)90579-9. [PubMed] [CrossRef] [Google Scholar]
22. Igoillo-Esteve M, Marselli L, Cunha DA, et al. Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia. 2010;53:1395–1405. doi: 10.1007/s00125-010-1707-y. [PubMed] [CrossRef] [Google Scholar]
23. Cnop M. Fatty acids and glucolipotoxicity in the pathogenesis of type 2 diabetes. Biochem Soc Trans. 2008;36:348–352. doi: 10.1042/BST0360348. [PubMed] [CrossRef] [Google Scholar]
24. Noushmehr H, D’Amico E, Farilla L, et al. Fatty acid translocase (FAT/CD36) is localized on insulin-containing granules in human pancreatic beta-cells and mediates fatty acid effects on insulin secretion. Diabetes. 2005;54:472–481. doi: 10.2337/diabetes.54.2.472. [PubMed] [CrossRef] [Google Scholar]
25. Carpentier A, Zinman B, Leung N, et al. Free fatty acid-mediated impairment of glucose-stimulated insulin secretion in nondiabetic Oji-Cree individuals from the Sandy Lake community of Ontario, Canada: a population at very high risk for developing type 2 diabetes. Diabetes. 2003;52:1485–1495. doi: 10.2337/diabetes.52.6.1485. [PubMed] [CrossRef] [Google Scholar]
26. Kashyap S, Belfort R, Gastaldelli A, et al. A sustained increase in plasma free fatty acids impairs insulin secretion in nondiabetic subjects genetically predisposed to develop type 2 diabetes. Diabetes. 2003;52:2461–2474. doi: 10.2337/diabetes.52.10.2461. [PubMed] [CrossRef] [Google Scholar]
27. Tushuizen ME, Bunck MC, Pouwels PJ, et al. Pancreatic fat content and beta-cell function in men with and without type 2 diabetes. Diabetes Care. 2007;30:2916–2921. doi: 10.2337/dc07-0326. [PubMed] [CrossRef] [Google Scholar]
28. Saisho Y, Butler AE, Butler PC. Pancreatic fat content and beta-cell function in men with and without type 2 diabetes: response to Tushuizen et al. Diabetes Care. 2008;31:e38. doi: 10.2337/dc08-0044. [PubMed] [CrossRef] [Google Scholar]
29. Saisho Y, Butler AE, Meier JJ, et al. Pancreas volumes in humans from birth to age one hundred taking into account sex, obesity, and presence of type-2 diabetes. Clin Anat. 2007;20:933–942. doi: 10.1002/ca.20543. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
30. Lee Y, Hirose H, Ohneda M, Johnson JH, McGarry JD, Unger RH. Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte–beta-cell relationships. Proc Natl Acad Sci USA. 1994;91:10878–10882. doi: 10.1073/pnas.91.23.10878. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
31. Lee Y, Lingvay I, Szczepaniak LS, Ravazzola M, Orci L, Unger RH. Pancreatic steatosis: harbinger of type 2 diabetes in obese rodents. Int J Obes (Lond) 2010;34:396–400. doi: 10.1038/ijo.2009.245. [PubMed] [CrossRef] [Google Scholar]
32. Morgan NG, Dhayal S, Diakogiannaki E, Welters HJ. The cytoprotective actions of long-chain mono-unsaturated fatty acids in pancreatic beta-cells. Biochem Soc Trans. 2008;36:905–908. doi: 10.1042/BST0360905. [PubMed] [CrossRef] [Google Scholar]
33. Erion DM, Shulman GI. Diacylglycerol-mediated insulin resistance. Nat Med. 2010;16:400–402. doi: 10.1038/nm0410-400. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
34. Perseghin G, Bonfanti R, Magni S, et al. Insulin resistance and whole body energy homeostasis in obese adolescents with fatty liver disease. Am J Physiol Endocrinol Metab. 2006;291:E697–E703. doi: 10.1152/ajpendo.00017.2006. [PubMed] [CrossRef] [Google Scholar]
35. Gastaldelli A, Cusi K, Pettiti M, et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology. 2007;133:496–506. doi: 10.1053/j.gastro.2007.04.068. [PubMed] [CrossRef] [Google Scholar]
36. D’Adamo E, Cali AM, Weiss R, et al. Central role of fatty liver in the pathogenesis of insulin resistance in obese adolescents. Diabetes Care. 2010;33:1817–1822. doi: 10.2337/dc10-0284. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
37. Tiikkainen M, Bergholm R, Vehkavaara S, et al. Effects of identical weight loss on body composition and features of insulin resistance in obese women with high and low liver fat content. Diabetes. 2003;52:701–707. doi: 10.2337/diabetes.52.3.701. [PubMed] [CrossRef] [Google Scholar]
38. Kirk E, Reeds DN, Finck BN, Mayurranjan SM, Patterson BW, Klein S. Dietary fat and carbohydrates differentially alter insulin sensitivity during caloric restriction. Gastroenterology. 2009;136:1552–1560. doi: 10.1053/j.gastro.2009.01.048. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
39. Guidone C, Manco M, Valera-Mora E, et al. Mechanisms of recovery from type 2 diabetes after malabsorptive bariatric surgery. Diabetes. 2006;55:2025–2031. doi: 10.2337/db06-0068. [PubMed] [CrossRef] [Google Scholar]
40. Morinigo R, Lacy AM, Casamitjana R, Delgado S, Gomis R, Vidal J. GLP-1 and changes in glucose tolerance following gastric bypass surgery in morbidly obese subjects. Obes Surg. 2006;16:1594–1601. doi: 10.1381/096089206779319338. [PubMed] [CrossRef] [Google Scholar]
41. Rodieux F, Giusti V, D’Alessio DA, Suter M, Tappy L. Effects of gastric bypass and gastric banding on glucose kinetics and gut hormone release. Obesity (Silver Spring) 2008;16:298–305. doi: 10.1038/oby.2007.83. [PubMed] [CrossRef] [Google Scholar]
42. Knop FK. Resolution of type 2 diabetes following gastric bypass surgery: involvement of gut-derived glucagon and glucagonotropic signalling? Diabetologia. 2009;52:2270–2276. doi: 10.1007/s00125-009-1511-8. [PubMed] [CrossRef] [Google Scholar]
43. Rubino F, Gagner M, Gentileschi P, et al. The early effect of the Roux-en-Y gastric bypass on hormones involved in body weight regulation and glucose metabolism. Ann Surg. 2004;240:236–242. doi: 10.1097/01.sla.0000133117.12646.48. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
44. Isbell JM, Tamboli RA, Hansen EN, et al. The importance of caloric restriction in the early improvements in insulin sensitivity after Roux-en-Y gastric bypass surgery. Diabetes Care. 2010;33:1438–1442. doi: 10.2337/dc09-2107. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
45. Laferrere B, Teixeira J, McGinty J, et al. Effect of weight loss by gastric bypass surgery versus hypocaloric diet on glucose and incretin levels in patients with type 2 diabetes. J Clin Endocrinol Metab. 2008;93:2479–2485. doi: 10.1210/jc.2007-2851. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
46. He S, McPhaul C, Li JZ, et al. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem. 2010;285:6706–6715. doi: 10.1074/jbc.M109.064501. [PMC free article] [PubMed] [CrossRef] [Google Scholar]