

 English
English Bahasa Melayu
Bahasa Melayu Bahasa Indonesia
Bahasa Indonesia Tiếng Việt
Tiếng Việt ไทย
ไทย
本翻譯僅作學術交流用,無商業意圖,請勿轉載,如有疑議問請來信
研究提出「果糖生存假說」,指出肥胖和代謝性疾病可能源於進化生物學的「生存開關」過度激活。這種反應由果糖攝入或內源性生產引發,導致飢餓、增重、脂肪積聚、胰島素抵抗等。研究表明,過度的果糖代謝不僅與肥胖相關,也可能促進糖尿病、高血壓等多種疾病的發生,並為肥胖的新療法提供了思路。
果糖生存假說與肥胖症
The fructose survival hypothesis for obesity
Johnson Richard J., Lanaspa Miguel A., Sanchez-Lozada L. Gabriela, Tolan Dean, Nakagawa Takahiko, Ishimoto Takuji, Andres-Hernando Ana, Rodriguez-Iturbe Bernardo and Stenvinkel Peter 2023The fructose survival hypothesis for obesityPhil. Trans. R. Soc. B3782022023020220230
https://royalsocietypublishing.org/doi/10.1098/rstb.2022.0230
Abstract
The fructose survival hypothesis proposes that obesity and metabolic disorders may have developed from over-stimulation of an evolutionary-based biologic response (survival switch) that aims to protect animals in advance of crisis. The response is characterized by hunger, thirst, foraging, weight gain, fat accumulation, insulin resistance, systemic inflammation and increased blood pressure. The process is initiated by the ingestion of fructose or by stimulating endogenous fructose production via the polyol pathway. Unlike other nutrients, fructose reduces the active energy (adenosine triphosphate) in the cell, while blocking its regeneration from fat stores. This is mediated by intracellular uric acid, mitochondrial oxidative stress, the inhibition of AMP kinase and stimulation of vasopressin. Mitochondrial oxidative phosphorylation is suppressed, and glycolysis stimulated. While this response is aimed to be modest and short-lived, the response in humans is exaggerated due to gain of ‘thrifty genes’ coupled with a western diet rich in foods that contain or generate fructose. We propose excessive fructose metabolism not only explains obesity but the epidemics of diabetes, hypertension, non-alcoholic fatty liver disease, obesity-associated cancers, vascular and Alzheimer’s dementia, and even ageing. Moreover, the hypothesis unites current hypotheses on obesity. Reducing activation and/or blocking this pathway and stimulating mitochondrial regeneration may benefit health-span.
This article is part of a discussion meeting issue ‘Causes of obesity: theories, conjectures and evidence (Part I)’.
摘要
果糖生存假說提出,肥胖和代謝性疾病可能是由於進化基礎生物學反應(生存開關)過度刺激而產生,該反應旨在在危機之前保護動物。這種反應的特徵是飢餓、口渴、覓食、體重增加、脂肪積聚、胰島素抵抗、系統性炎症和血壓升高。這個過程是由攝入果糖或通過多醇途徑刺激內源性果糖產生來啟動的。與其他營養素不同,果糖降低了細胞中的活性能量(三磷酸腺苷),同時阻止了其從脂肪儲存中再生。這是由細胞內尿酸、粒線體氧化應激、AMP激酶的抑制和抗利尿激素的刺激介導的。粒線體氧化磷酸化被抑制,糖酵解被刺激。盡管這種反應旨在是溫和且短暫的,但由於‘節儉基因’的獲得與富含含有或產生果糖的食物的西方飲食,人類的反應過度。我們提出過多的果糖代謝不僅解釋了肥胖,還解釋了糖尿病、高血壓、非酒精性脂肪肝病、與肥胖相關的癌症、血管和阿爾茨海默病的流行病學,甚至衰老。此外,該假說結合了關於肥胖的當前假設。減少激活和/或阻止此途徑,並刺激粒線體再生,可能有益於健康壽命。
本文是“肥胖的原因:理論、猜測和證據(第一部分)”討論會議問題的一部分。
1 簡介
科學發現中常常被忽視的一種方法是探討大自然及其伴隨的進化機制如何找到解決棘手問題的方法 [1,2]。這種仿生學的方法可以提供關於野生動物在克服逆境時使用的巧妙方法的見解,為基於這些方法的新療法提供啟示 [3,4]。然而,進化也可能無意中導致疾病。事實上,有人提出,在一個資源稀缺的世界中幫助生存的遺傳適應(節儉基因)在一個充裕的世界中可能會“逆向作用”,增加肥胖和糖尿病的風險 [5]。
在這裡,我們討論了一個最近發現的保護機制,我們稱之為“生存開關”,它在資源變得匱乏之前啟動。我們的研究表明,這種效應是由果糖介導的,不同於葡萄糖的主要生物學功能是提供即時能量,果糖的主要功能是幫助儲存能量。這些不同的生物學功能是由葡萄糖和果糖代謝如何調節細胞內能量水平的結果。我們討論了果糖的多種來源以及它如何介導其生物學作用。我們還提出兩個事件導致了這個保護途徑轉變為引發疾病的途徑。第一個事件是獲得了“節儉基因”(或更準確地說是創建了節儉基因型的基因的損失),第二個事件是含有或產生果糖的食物大幅增加。這兩個事件導致了我們所提出的“生存開關”的過度激活,我們認為這是導致肥胖和許多今天影響我們的“生命負擔”非傳染性疾病的原因。
2 果糖的來源,生存開關的觸發器
果糖是一種簡單的糖,是水果和蜂蜜中的主要營養成分。然而,在西方飲食中,它的主要來源是蔗糖,它由果糖和葡萄糖結合在一起,以及高果糖玉米糖漿(HFCS),它由果糖和葡萄糖的混合物組成,通常果糖的濃度稍高,因為研究表明人類更喜歡稍微更多果糖,因為它比葡萄糖更甜。今天,這些“添加糖”佔整體能量攝入的約15%,有些人群攝入的量甚至高達20%或更多 [6]。
果糖也可以從葡萄糖在體內產生(圖1)。這發生在葡萄糖水平(即底物)過多的情況下,例如糖尿病、攝入高升糖碳水化合物後以及高碳水化合物飲食 [7–9]。葡萄糖轉化為果糖的酶反應稱為多醇途徑,限速酶是醛酮還原酶(AR)。 AR受壓力的激活,例如脫水(高滲透壓)、飢餓(例如缺血)或低氧環境。鹹食和酒精(都會提高血清滲透壓)以及果糖本身也會刺激果糖的生成 [7,10,11]。富含味精和核苷酸(如谷氨酸和腺苷一磷酸(AMP)和次黃嘌呤一磷酸(IMP))的鮮味食品也會生成尿酸,刺激果糖的產生 [12,13]。所有這些營養素都會刺激肝臟中的果糖生成,足以完全激活生存開關 [14]。果糖的生成也可以在其他器官中刺激,如大腦、腎臟、血管和心臟。實際上,高血糖時,大腦內會產生果糖 [9],心臟缺血時會產生 [15],腎臟缺血再灌注損傷時會產生 [16],裸鼴鼠在穿越低氧巢穴時會在循環中產生 [17],並且在胎盤和胎兒在完全形成胎盤循環之前的第一季度缺氧期間也會產生 [18]。酒精由於提高滲透壓的能力,也可以刺激果糖的產生 [11]。
圖1{kind=link}
果糖改變體重調節。在正常情況下,大多數動物會嚴格調節其體重。如果它們被餵食高卡路里飲食,它們將增加體重,如果它們被給予低卡路里飲食,則將減輕體重,但當它們被允許恢復正常飲食時,它們將自動調整到正常體重。相反,果糖通過降低細胞內ATP的同時阻礙了從脂肪儲存中補充ATP。在幾天到幾周的時間內,動物會產生瘦素抵抗,導致能量攝入增加。然而,由於氧化應激的抑制,ATP的產生保持在低水平。因此,攝入的熱量優先用於產生脂肪。隨著時間的推移,ATP水平會恢復,但以極大的增加脂肪儲存為代價。(在線顏色版本。)
雖然大多數評估內源性果糖產生的研究是在實驗室動物中進行的,但有數據表明,年輕的瘦身成年人體內內源性果糖的產量可能超過5克/天,在攝入高升糖軟飲料後,果糖產生率可能增加三倍或更多 [8]。在攝取高升糖、高鹽或高糖飲食的受試者中,預計產量會更高,因為已知在這種情況下醛酮還原酶(AR)的表達會上調。高升糖和高鹽飲食也可以在大腦中誘導明顯增加的果糖生成 [9,10]。因此,果糖可以從飲食中獲得和/或生成(糖、高果糖玉米糖漿、高升糖碳水化合物、鹹食、鮮味食品、酒精),同時也可以在壓力條件下(缺血、低氧和脫水)產生。事實上,三種吸引人的味道(甜味、鹹味、鮮味)都鼓勵攝取生成果糖的食物 [7,10,12,19],而苦味和酸味可能是為了避免攝取可能攜帶毒素的食物而發展的。
3 果糖通過降低ATP水平來觸發生存開關
體重通常受到嚴格調節,特別是對於野外的動物來說 [20,21]。其中一個最強的調節因子似乎是瘦(無脂肪)體重,這個測量與能量攝入和靜息能量代謝都有關 [22]。肌肉質量是一個動物試圖保護的關鍵特徵,反過來受到細胞色體功能水平的指導 [23]。因此,動物試圖保持細胞內ATP水平,因為這代表了它們的活性能量,它們通過在代謝靈活性中擁有的方式來實現這一點,其中任何耗盡的ATP都可以迅速從營養攝入或脂肪儲存生成的ATP進行替換(圖1)。在這種情況下,對於不會在能量支出上急劇增加的超卡路里飲食的施行將導致體重增加(和脂肪積累),而低卡路里飲食則將導致體重減輕(和脂肪儲存的耗盡)。在這兩種情況下,能量平衡保持不變,並保持ATP水平。
相比之下,果糖代謝方式不同,儘管它也遵循能量平衡的規則。具體來說,果糖主動降低細胞內ATP,同時降低了生成新ATP的能力(圖1)。因此,代謝靈活性被阻塞。ATP水平不會下降到威脅生存的程度,但足以觸發警報,可用能量儲存可能會被耗盡。例如,在人類的研究中,經口攝入果糖後,肝臟的ATP水平可以下降20% [24],如果靜脈內給予,則可以下降60-70% [25]。ATP耗盡的程度與肝臟暴露於果糖的濃度有關,這與攝入量和吸收速度有關,當果糖以液體形式給予時,吸收速度更快 [26,27]。這觸發了一系列生物學反應,導致能量攝入增加。然而,持續抑制細胞色體功能導致卡路里被轉移到儲存能量(脂肪)。最終,ATP水平得到替換,但以存儲更多脂肪的代價,從而使整體能量水平(即活性和儲存能量)更高。
果糖降低細胞內ATP的具體機制顯示在圖2中。果糖迅速被果糖激酶C(也稱為酮己糖激酶C或KHK-C)磷酸化為果糖-1-磷酸,該酶沒有反饋系統來保護ATP水平,因此細胞內ATP和磷酸急劇下降 [28,29]。細胞內磷酸的下降刺激AMP脫氨酶-2(AMPD2),它去除AMP底物 [29],從而減緩了ATP的再生,同時刺激尿酸的產生(既從AMP降解為IMP,也從新的嘌呤合成中)[30,31]。這有助於維持(AMP + ADP)/ATP的正常電荷比。儘管一些IMP可能潛在地可以通過嘌呤救助途徑轉化回AMP和ATP [32],但似乎更有利於尿酸的產生,攝取後的第一個小時內,血清尿酸可以增加0.3-2.0 mg dl−1 [30,33,34]。
圖2{kind=link}
內源性果糖的生成。果糖是從葡萄糖(其底物)通過多醇途徑生成的。醛酮還原酶是限速酶。促使果糖生成的主要因素是高血糖水平,以及可以刺激醛酮還原酶活性的因素。從食物的角度來看,這包括高升糖碳水化合物、鹹食、酒精和啤酒等鮮味食品。然而,這條途徑也會被壓力激活,包括缺血、低氧、脫水和熱壓力。(在線顏色版本。)
由黃嘌呤氧還原酶生成尿酸會產生氧化劑(主要是過氧化氫)[35],但尿酸本身也刺激NADPH氧化酶[36–40],該酶遷移到細胞色體[41]。從NADPH氧化酶和內源性細胞色體氧化應激兩方面,以及保護性抗氧化系統(尤其是Nrf2)的同時減少,導致了細胞色體的氧化應激增加[41,42]。氧化應激通過抑制柠檬酸循環中的aconitase[41,43–45],以及通過乙酰化肉碱棕櫚酰轉移酶-1α(參與脂肪酸運入細胞色體的酶)[46]和乙烯基CoA水合酶(β脂肪酸氧化途徑中的酶)[43,47]來阻止β脂肪酸氧化,並通過AMP活化的蛋白激酶[47,48]來阻止ATP再生。aconitase的抑制會刺激參與脂肪生成的酶[41,49],而腸道微生物對果糖的反應產生醋酸,用於生成提供底物的乙酰CoA。此外,有證據表明,果糖代謝可以導致煙酰胺腺嘌呤二核苷酸(NAD+)的消耗,降低NAD+/NADH比值,影響氧化還原平衡,並導致去乙酰酶的減少,這也可能帶來代謝效應[42,50],並增強糖酵解反應[51]。這可能是導致CPT1α乙酰化的原因[46]。
儘管細胞色體氧化磷酸化和ATP生成受到抑制,果糖-1-磷酸的生成刺激了葡萄糖激酶從細胞核釋放出來,導致葡萄糖攝取和糖原生成,而果糖-1-磷酸的分解生成了甘油醛和二羥基乙酮磷酸。在禁食狀態下,這會刺激葡萄糖生成作為能量底物的葡萄糖生成[52],而在進食狀態下,優先刺激糖酵解[53]。例如,根據放射示踪研究,乳酸可以佔攝入果糖的25% [54]。儘管乳酸可以用於生成作為柠檬酸循環底物的乙酰CoA,但當乳酸積聚時,它會生成活性氧自由基,損害脂肪酸進入細胞色體的能力,並減少細胞色體ATP生成[55]。果糖通過刺激糖酵解的淨刺激[28,41,47,48],同時抑制氧化磷酸化的氧消耗,可能旨在為潛在缺氧的動物提供生存利益[17]。
降低細胞內ATP的這一策略似乎是啟動生存反應並破壞體重調節的核心所在。實際上,卡路里攝入受到刺激以補償ATP不足,但這一轉變將卡路里轉移到脂肪。最終,ATP水平得到恢復,但代價是增加脂肪含量。隨著時間的推移,由於反復氧化應激對細胞色體的作用導致永久性細胞色體功能異常和消耗[43,56,57],存在一種過渡。現在,ATP水平始終保持較低,但人體適應了低ATP水平,體脂肪質量減少,靜息能量代謝下降。能量攝入必須保持低水平,否則將發生體重回升。
與減少細胞內ATP是肥胖和代謝綜合症的重要觸發因素的假設一致,果糖[16,58–61]和尿酸[12,43,44,62]可以在各種細胞類型和組織中誘導低ATP水平。果糖誘導的肝臟ATP耗竭可以通過安乐脉噻嗪部分逆轉[45]。更重要的是,低細胞內ATP狀態是肥胖、糖尿病、非酒精性脂肪肝病(NAFLD)和阿爾茨海默病的特徵[25,63–66]。
4 由果糖代謝誘導的「生存開關」描述
果糖的投入可以完全複製代謝綜合症,並導致體重增加、內臟脂肪積累、胰島素抵抗、高三酸甘油脂血症、低高密度脂蛋白膽固醇、高血壓、脂肪肝、微量白蛋白尿、高尿酸血症以及全身炎症標誌物(包括低脂肪細胞素、高瘦素和高敏感性C-反應蛋白升高)[67]。這些發現也在準備冬眠的動物中觀察到,這表明術語「代謝綜合症」可能是一個不當名稱,而這些特徵應該被描述為「脂肪積儲綜合症」[68]。所有這些特徵都是作為生存反應的一部分發展的(圖3),具體描述如下:
圖3{kind=link}
果糖代謝和降低細胞內ATP水平的機制。果糖首先由KHK-C磷酸化為果糖-1-磷酸,導致ATP的迅速消耗,該消耗直接依賴於果糖的濃度。與葡萄糖不同,其ATP從未大幅減少,KHK-C驅動的反應可以使ATP和細胞內磷酸濃度暴跌。反過來,低細胞內磷酸會觸發AMP脫氨酶-2(AMPD2)的激活。在ATP枯竭過程中產生的AMP然後被代謝為次黃嘌呤單磷酸(IMP),最終生成尿酸。使用AMP會去除一些再生ATP所需的AMP底物。然而,細胞內尿酸刺激NADPH氧化酶,該酶遷移到細胞色體,同時抑制了細胞色體的抗氧化保護機制,尤其是Nrf-2。細胞色體的氧化應激抑制了細胞色體β脂肪酸氧化(通過抑制乙烯基CoA水合酶)和三羧酸循環(通過阻止aconitase),從而抑制了氧化磷酸化產生ATP。此外,尿酸還抑制了AMP活化的蛋白激酶(AMPK),該酶有助於在低能量狀態下再生ATP。因此,果糖代謝KHK-C的淨效應是降低細胞內ATP水平。(在線顏色版本。)
(a) 尋找食物和水
果糖鼓勵行為變化以幫助尋找食物和水。這包括通過激活促進飢餓和狂食行為的促進飢餓激素-下視丘回路,儘管這本身並不刺激增加的食物攝入量[69,70]。果糖還刺激口渴,可能是通過由於水轉移到細胞中與糖原生成相關而增加的血清滲透壓[71]。最重要的是,果糖通過損害飽足感干擾了正常的體重調節,導致過量食物(能量)攝入。這需要幾個星期才能發展,並且是由中央(下視丘)瘦素抵抗所介導的[72,73]。果糖還作用於大腦,刺激尋找反應,包括刺激探索行為、冲動性和增加的運動活動[74,75]。通過提高尿酸也可以觀察到類似的效應[74,75]。研究表明,這些效應是通過抑制大腦中與自我控制、近期記憶和深思熟慮相關的對胰島素敏感區域所介導的,如大腦皮質、嗅皮質、海馬和後扣帶皮質[76,77]。
(b) 增加脂肪和糖原儲存
除了通過誘導瘦素抵抗[72,73]來刺激過量卡路里攝入外,果糖還增加了腸絨毛的長度,可能有助於更有效地吸收食物[78]。果糖代謝還刺激了脂肪生成[38,41,79],損害了β脂肪酸氧化[57,80],並且可能由於高胰島素血症的發展而減少了脂肪細胞的脂解作用[81]。糖原儲存也增加[82–84]。
(c) 能量保留
儘管尋找反應需要能量支出,但這是由於休息能量代謝的下降而得到補償,這可能與減少的代謝有關,次要原因包括脂肪酸氧化的阻塞以及胰島素抵抗對肌肉中葡萄糖代謝的影響[85]。此外,骨骼肌和脂肪細胞對葡萄糖的減少攝取結果是保留血糖水平,這可能保護了那些對胰島素對葡萄糖攝取不太依賴的大腦區域,這些區域的血糖水平可能保持正常或高,[86]。儘管如此,一些大腦區域對胰島素更依賴,特別是那些參與抑制尋找反應的區域[74]。果糖損害了與自我控制、深思熟慮和近期記憶[76,77]有關的區域的胰島素信號傳遞[87]。結果是尋找的刺激,而能量保留則由全身和大腦胰島素抵抗[75,88]共同協助。
(d) 關鍵體功能的保護
處於危險狀況的動物需要保持強健的循環和排泄功能。果糖代謝導致急性血壓升高,這是依賴尿酸的[89,90],這並不奇怪。通過增加腎小球過濾壓,可以保持腎功能,實驗性高尿酸血症也觀察到類似的發現[91,92]。果糖還刺激抗利尿激素加壓素的產生[93,94],這可以幫助重吸收水分,果糖還刺激了腎臟近曲小管[95,96]和腸道[97]中的鈉吸收。這些效應有助於生存(圖4)。
圖4{kind=link}
對果糖的生存反應
(e) 免疫系統的啟動
果糖生成的尿酸與炎症途徑的刺激相關,包括MAP激酶(p38)、NF-ΚB的活化以及炎症體的刺激[98],還有助於樹突狀細胞功能[99,100]。升高的尿酸刺激了氧化壓力[37,101]以及趨化因子、C-反應蛋白和炎症途徑的產生[102,103]。再次,免疫啟動提供了免受感染的保護。
(f) 進入低功耗模式
果糖代謝引起的葡萄糖氧化磷酸化減少並刺激糖酵解,使身體進入低功耗模式,降低了其氧氣需求[17]。這在局部缺血性炎症區域提供了初始好處,而在長期內可能會促使炎症和纖維化。例如,心臟缺血時,會產生果糖,並抑制細胞色素氧化磷酸化和刺激糖酵解,這促使心臟重塑[104]。這似乎部分依賴於轉錄因子HIF-1α的活化[104,105]。受刺激的糖酵解和降低的細胞色素氧化磷酸化活性也是糖尿病性腎病的特徵[106,107]。我們報導了糖尿病性腎病是由於在腎臟中將葡萄糖轉化為果糖而介導的[108]。SGLT2抑制劑在心臟和腎臟疾病中的效益可能是通過阻止疾病組織中的葡萄糖攝取,從而防止其後續轉化為果糖[109]。果糖代謝的阻礙可能啟動脂肪氧化和AMP-活化蛋白激酶活性,類似於休眠動物中觀察到的現象[80,110]。果糖減少細胞色素氧化磷酸化並刺激糖酵解(華爾堡效應)的效應解釋了為什麼癌細胞更喜歡果糖作為生長媒介[111],以及為什麼癌細胞生長受到果糖和尿酸的刺激[112,113]。
5 果糖代謝在肥胖和代謝綜合症流行病中的作用
肥胖和糖尿病的流行病在二十世紀初開始出現[114,115]。一個可能的機制與糖的攝取急剧增加有關,當高果糖玉米糖漿(HFCS)引入時,出現急劇的轉折[115,116]。含果糖的軟飲料和其他含有果糖的液體特別有效地激活了這條途徑,因為ATP的下降與肝細胞暴露的濃度直接相關,這不僅與果糖的量有關,還與攝入速度有關[26]。事實上,我們發現減緩攝取含果糖的蘋果汁可以減少生存途徑的激活[27]。事實上,含糖飲料的攝取與肥胖和代謝綜合症的風險相關。同樣,全球肥胖和糖尿病的流行病與糖的攝取量增加以及糖在非西方文化中的引入相關[117]。此外,攝取富含糖和鹽分的加工食品以及攝取高升糖指數碳水化合物和酒精也有助於攝取的果糖量或內源性生成以及肥胖、2型糖尿病和高血壓的風險[118]。
然而,有證據表明,相對於大劑量的給藥,人類對果糖更為敏感,因為老鼠和大鼠通常對果糖的影響相對抵抗,除非給予大劑量。在這方面,我們的團隊發現了兩種明顯增加果糖誘導代謝綜合症風險的基因突變。第一個是維生素C的突變,大約在約6100萬年前發生[119],而這個時間相對靠近結束白堊紀時期的小行星撞擊事件[120]。維生素C是一種可以阻止果糖作用的抗氧化劑,已被發現對代謝綜合症的各個組成部分具有有益作用[121]。我們的團隊發現,餵食果糖的維生素C缺乏小鼠呈現一種劑量反應,其中較高劑量的維生素C對於攝入相同量的果糖來說導致的肥胖較少(未發表觀察)。因此,維生素C的突變可能為早期靈長類在小行星碰撞後的“撞擊冬季”期間努力生存提供了自然選擇優勢[122]。
另一個可能有益於我們靈長類祖先的突變是尿酸酶突變,從大約2400萬年前開始分步進行,直到基因在中中新世完全滅絕。尿酸酶是一種分解尿酸的酶,而缺乏尿酸酶的小鼠對果糖的反應中尿酸升高劇烈增加[123]。我們已經討論了失去尿酸酶的證據可能代表了一個真正的“節約基因”[124]。這種突變發生在原始大猿幾乎從全球變冷中滅絕的時期[125]。有關它可能是一個節約基因的證據表明,抑制尿酸酶可以顯著增強果糖誘導大鼠代謝綜合症的能力[126],而復活原始的尿酸酶已被證明可以阻止果糖對人類肝細胞的脂肪生成影響[127]。值得注意的是,當這種突變發生時,它只使血清尿酸水平增加一倍(約為3-4 mg dl-1),因此主要是為了防止饑餓而不是引發肥胖[128]。然而,在二十世紀,血清尿酸水平隨著糖攝入量的增加和肥胖、糖尿病和心血管疾病的增加而急剧上升[128]。血尿酸水平大於8 mg dl-1的人更有可能患肥胖和代謝綜合症,實驗和初步研究表明,尿酸可能起著一個促進作用[124]。
6 果糖如何導致體重增加?
觀察到果糖既刺激食物攝入又降低靜息能量代謝,這表明增加的能量攝入和降低的能量消耗都可能導致肥胖,這是通過體重和脂肪質量的增加來確定的。然而,靜息能量消耗的減少部分被覆蓋在覓食反應中使用的能量增加。
為了評估這兩種機制的重要性,我們進行了成對餵養研究,即給予大鼠高果糖(有時也含糖)食物,然後與具有相同組成和卡路里含量的食物進行比較,唯一不同的是果糖被澱粉取代[129–132]。在某些研究中,動物被限制攝取熱量,但每組攝取的熱量相等[132]。主要發現是,攝入相同熱量的動物,無論飲食是否含果糖,體重變化相似。雖然給予含糖或果糖的動物體重稍微增加的趨勢(這可能是由於靜息能量代謝降低所致),但總體體重變化不顯著[132]。因此,體重增加主要是由增加的熱量攝入所解釋的[129–132],至少在持續四個月或更短時間的研究中是這樣。
為了找出導致能量攝入增加的原因,我們進行了額外的研究。首先,我們發現甜味可以促進果糖攝入,但缺乏味覺的老鼠仍然喜歡果糖而不是水,並且隨著時間的推移變得肥胖(比對照組更為肥胖)[19]。對果糖的偏好取決於果糖的代謝,因為缺乏果糖激酶的老鼠對果糖幾乎沒有喜好,盡管他們仍然喜歡蔗糖,這可能是由於其中含有的葡萄糖[19,133]。重要的是,對果糖的偏好並不是導致熱量攝入增加的原因,這是基於我們在特定器官中敲除果糖激酶的研究結果。事實上,對果糖的偏好是由腸道中的果糖激酶介導的,而代謝綜合症是由肝臟中的果糖代謝驅動的。重要的是,僅在肝臟特定部位缺乏果糖激酶的老鼠會飲用大量果糖,但完全免受體重增加和代謝綜合症的影響[14]。
增加能量攝入的主要機制是瘦素抵抗的形成。最初,食用蔗糖或果糖的動物會減少飼料攝入,以維持中性能量平衡,以防止體重增加。然而,幾個星期後,動物增加了飼料攝入,使總熱量攝入變得高熱量,並且這與體重增加相關。研究表明,這是由於中樞瘦素抵抗的形成,並且可以通過阻止果糖代謝來預防[72,73]。事實上,我們發現,這一機制是由果糖激酶C亞型專門介導的,並且是由於副產物反應驅動的能量耗盡[129,134]。
準備進入冬眠的動物表現出與生存開關相似的特徵(例如,暴飲暴食、瘦素抵抗、體重增加和脂肪積聚、脂肪肝和胰島素抵抗),但在即將進入冬眠之前,它們會減少進食,即使食物仍然可用,這與它們的新陳代謝下降有關[135,136]。可能的一個機制與達到觸發減少進食的生物學反應(重力調節器)[137,138]的體重有關。盡管這種機制仍然存在於肥胖的人類中[139],但顯然它可能會被擊敗,我們猜測這可能與持續暴露於高濃度的果糖有關。
(a)脂肪的作用
瘦素抵抗似乎是由攝入或內源性果糖引起的,而其他食物,如高脂肪飲食,不會引起瘦素抵抗[72,73]。儘管果糖介導了瘦素抵抗,但在變得對瘦素抵抗的動物中攝入高能量密度、高脂肪的食物會增加體重[72,73]。
事實上,阻礙瘦素信號傳遞會刺激對高脂肪食物的偏好[140],這是有道理的,因為它會促使體重增加和脂肪積聚更快。地松鼠在冬眠前積極增重時會優先食用富含高多不飽和脂肪的種子[141]。此外,最近研究表明,由TRPM5味覺受體[142]以及促使對脂肪偏好的腸道迷走神經通路調節的脂肪味覺存在[143]。這就是為什麼脂肪與果糖或糖的結合會比單獨攝取脂肪或果糖更大幅度地增加脂肪積聚和體重增加的原因之一[144]。此外,西方飲食的人們可能攝取足夠的糖、鹽、高升糖碳水化合物和酒精,以在大多數個體中誘導一些瘦素抵抗。例如,我們報導過,內源性果糖可以在餵養類似西方飲食(50%碳水化合物)的小鼠的情況下誘導老化變化,儘管糖含量較低-缺乏果糖激酶的小鼠保持瘦且健康[145]。這些發現還解釋了為什麼因紐特人儘管攝取高脂肪、高蛋白質飲食仍保持苗條[146],以及為什麼“低碳水化合物”飲食不會引起體重增加,儘管脂肪含量高。例如,在正常小鼠中,豬油不會引起體重增加,除非首先用果糖使其對瘦素產生抵抗[73]。
(b)葡萄糖的作用
我們攝取的大部分果糖來自於含有葡萄糖的添加糖(蔗糖和高果糖玉米糖漿,HFCS),這些葡萄糖可以明顯增強果糖的吸收,從而加重果糖的影響[147,148]。然而,還有證據表明,單獨給予葡萄糖也可能誘導肥胖和代謝綜合症[7]。一個流行的假設是,添加糖和高升糖碳水化合物可能通過過度刺激胰島素[149]來引起肥胖,胰島素不僅刺激脂肪的儲存,還阻止了脂肪細胞中的脂解[81]。然而,正如前面提到的,高升糖食物也可以激活多醇途徑並生成內源性果糖[7,14]。
為了評估葡萄糖-胰島素軸在驅使肥胖中的作用,我們將葡萄糖給予KHK-KO小鼠或對照小鼠[7]。對照小鼠體重急劇增加,並伴有脂肪肝和胰島素抵抗,但肝臟中也有高水平的果糖。缺乏KHK的小鼠喝相同量的葡萄糖,但進食較少,儘管它們發展出輕微的肥胖,但大約只有野生型對照組觀察到的一半。 KHK-KO小鼠還幾乎沒有脂肪肝或胰島素抵抗。因此,這些研究確認了葡萄糖很可能通過反复刺激胰島素來引起一些肥胖,但高升糖碳水化合物引起肥胖和代謝綜合症的主要機制之一是通過內源性生成果糖。稍後的研究進一步支持了這一觀點,當我們將HFCS給予缺乏KHK的小鼠時,發現它提供了更多的保護,以防止肥胖、脂肪肝和胰島素抵抗,葡萄糖-胰島素途徑的效應約為四分之一[14]。因此,盡管碳水化合物-胰島素模型仍然是引起肥胖的一個重要機制,但果糖的轉化在高升糖碳水化合物引起肥胖的過程中具有重要作用。有趣的是,果糖誘導的胰島素抵抗導致高空腹胰島素水平,因為脂肪細胞在胰島素抵抗狀態下仍對胰島素的抗脂分解作用敏感[81]。因此,果糖誘導的胰島素抵抗也支持了葡萄糖-胰島素假設,但有內源性果糖作為中間步驟。
(c) 蛋白質的作用
蛋白質對於保持瘦身體質量至關重要,有一些證據表明,低蛋白質飲食可能會刺激增加能量攝入以實現這一目標,但卻增加了體脂儲存(蛋白質利用假設)[150]。然而,大多數低蛋白質飲食都是高碳水化合物飲食,很難知道在驅使肥胖方面哪一個更重要,而且低蛋白質飲食還會刺激FGF21,它通過刺激脂肪的氧化,尤其是在肝臟中,來對抗果糖的作用[151]。
此外,某些蛋白質,如紅肉和加工肉,可能會增加患糖尿病、肥胖、痛風和慢性腎病的風險[152–157]。一種可能的解釋是,紅肉和貝類富含谷氨酸和核苷酸,如IMP和AMP [158]。IMP是果糖能量消耗途徑的關鍵組成部分,谷氨酸也在肝臟中代謝成尿酸。事實上,我們發現鮮味(包括谷氨酸和IMP)可以生成尿酸,引起肝臟ATP枯竭、白蛋白抵抗、肥胖和代謝綜合症,類似於果糖[12]。使用維諾博凍治療能夠阻止味精引起肥胖的效應[12]。谷氨酸刺激尿酸的機制可能與轉化為谷氨酰胺並新生尿酸有關[159,160]。已知,痛風患者往往有高血清谷氨酸水平,也因富含谷氨酸的食物(如番茄)而患痛風[161,162]。然而,大多數富含嘌呤的蔬菜似乎不太可能升高尿酸並引起痛風[163],這很可能是因為這些嘌呤相對較少含有AMP和IMP [158]。
有趣的是,升高尿酸的嘌呤和谷氨酸豐富的食物往往是帶有自己口味的鮮味食物,這表明這些是動物傾向於尋找的食物[164,165]。雖然尿酸通過其刺激線粒體氧化壓力並抑制AMPK的能力在直接激活開關方面發揮著作用[41,47],但它還可以刺激果糖的生成並增強KHK的表達[13,166]。因此,大部分效應是通過從葡萄糖生成果糖來刺激,這可能解釋了在沒有碳水化合物的情況下高蛋白質飲食不會引起肥胖的原因。
7 果糖與代謝綜合症:一種獨立於過多熱量的效應
我們的成對餵食研究中一個引人注目的效應是,儘管體重增加是由過多熱量引起的,果糖的其他效應在熱量攝入受到限制時仍然發生[129–131]。實際上,在一項實驗中,大鼠被餵以低熱量、高蔗糖飲食,蔗糖飲食的大鼠仍然出現了嚴重的脂肝、高三酸甘油脂血症、胰島素抵抗和血壓升高[132]。我們還發現,大鼠最初出現高胰島素血症,但血糖水平正常,隨著時間的推移,他們的胰島素逐漸下降,並伴隨著明顯的糖尿病的發展。胰島也顯示了透明質沉著和發炎,胰島細胞上新生表達了尿酸轉運蛋白。澱粉餵食的對照動物也出現了輕度高三酸甘油脂血症和胰島素抵抗的一些徵兆,我們現在認為這可能是來自低濃度果糖的產生。然而,這兩組之間的整體差異是顯著的[132]。這些研究證實,代謝綜合症,包括導致體脂增加的身體組成的改變,可以與體重增加很少或沒有關聯。實際上,非酒精性脂肪肝病可以發生在瘦削的人身上[167],尤其是在有高尿酸血症的情況下[41]。
8 鹽、脫水和抗利尿素在肥胖中的意外角色
水是一個關鍵的資源,果糖增加抗利尿素血液水平以增加對抗利尿素依賴的尿液濃縮,以保持水分。我們還發現,果糖在抗利尿素調節中可能具有基本作用,因為脫水(高滲透壓)被發現能誘導上視核中的多元醇途徑的激活,伴隨著抗利尿素合成的增加,而KHK-KO小鼠對急性和慢性脫水的抗利尿素反應較差[169]。
最令人驚訝的是,抗利尿素不僅僅刺激腎臟中的水重新吸收,而且它位於果糖下游,它主導了生存開關的大多數特徵。多年來,已經知道還有另一種抗利尿素受體,稱為V1b受體,但其生理功能尚不清楚。我們發現,V1b受體敲除小鼠完全免受果糖誘導的代謝綜合症的影響[93]。儘管具體機制尚不完全清楚,但我們知道V1b受體刺激腎上腺皮質激素(ACTH)和葡萄糖素水平,並且它可能還調節KHK的表達[93]。
回顧一下,已知海洋和沙漠哺乳動物使用脂肪作為代謝水的來源[170,171]。動物在冬眠、夏眠和長距離遷徙的鳥類期間依賴代謝水。攝入鹽可以誘導高滲透壓和一種類似脫水的狀態,而這也可以激活果糖和抗利尿素的產生,並導致實驗室小鼠中與果糖激酶相關的肥胖和代謝綜合症[10]。攝取鹽高的肥胖者預測代謝綜合症、糖尿病和脂肪肝的發生[10]。大多數肥胖者也表現出脫水的跡象,並具有高抗利尿素水平(通過測量血清copeptin來識別)[172–174]。用水來補充水分以抑制抗利尿素水平也可以部分地停止甚至逆轉代謝綜合症的特徵[10]。這些研究表明,生存開關與一種營養物質(果糖)、一種代謝廢物(尿酸)和一種激素(抗利尿素)密切相關,並且肥胖確實是一種激素失調。
9 生存開關的短期和長期健康後果
生存開關持續啟動的最嚴重後果之一,不僅僅是肥胖和體重增加,還有涉及西方社會常見疾病的代謝效應(表1)[16,18,25,38,41, 47,74,75,91,92,111,132,145,175–184]。這不僅包括與肥胖相關的經典疾病,如糖尿病、非酒精性脂肪肝和痛風,還包括高血壓、冠狀動脈疾病、某些癌症、行為障礙和失智症等疾病。
表格1生存開關的過度活化可能導致代謝性疾病。
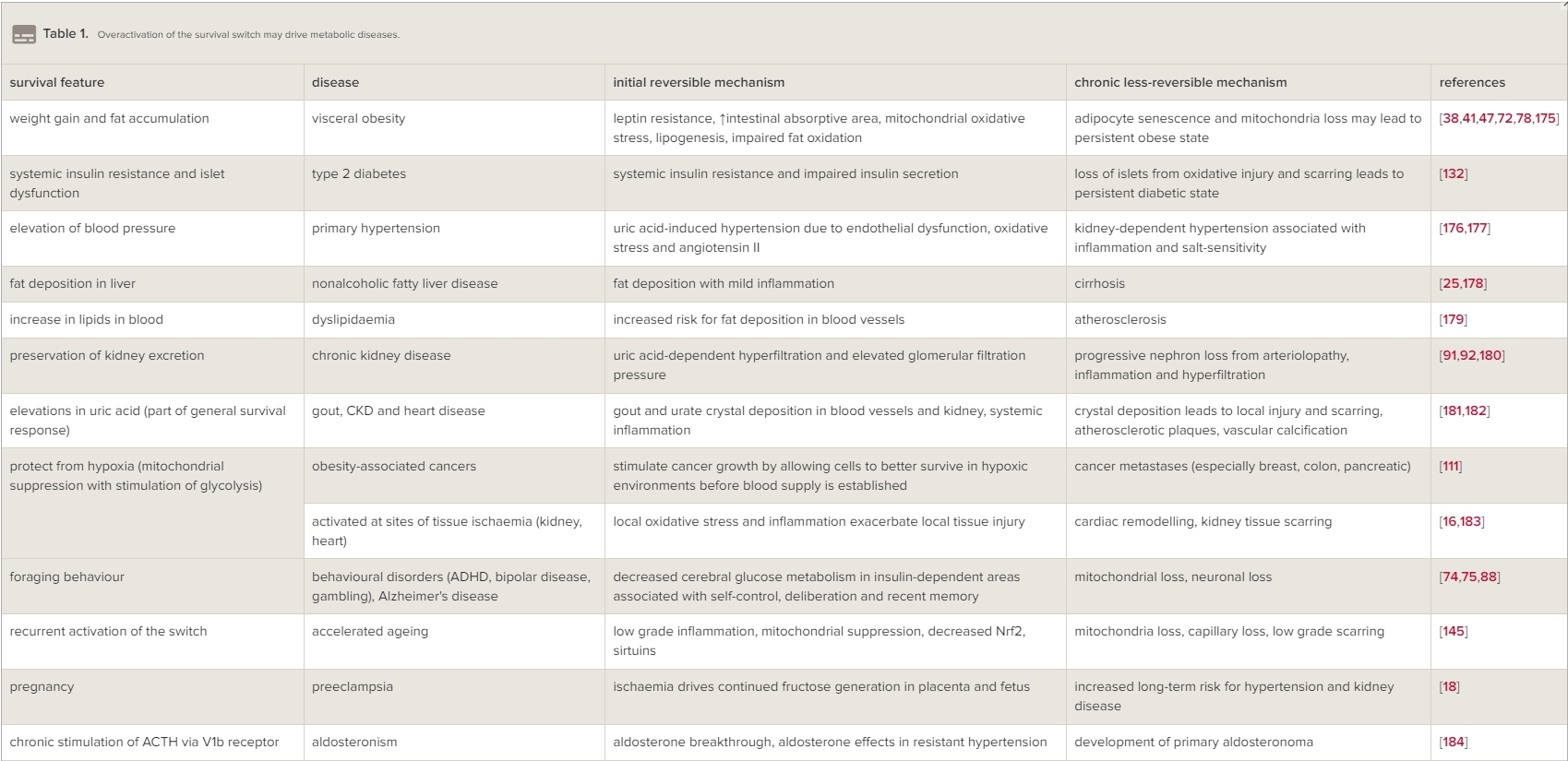
一個例子是認識到這一途徑在驅動冠心病方面的新興作用。近來,人們認識到系統性發炎以及動脈粥樣硬化斑塊中的發炎在冠心病和心肌梗死中起到了作用[185]。潛在的機制之一是可溶性尿酸的作用,可以激活發炎體和NFkB介導的發炎[98,102]。然而,最近發現了一個更戲劇性的機制,即85%的痛風患者在他們的血管中有尿酸結晶,似乎集中在動脈粥樣硬化斑塊中[181,186]。事實上,高尿酸血症和痛風與心血管死亡風險增加有關,這是由流行病學研究和Mendelian隨機化研究確定的[187,188]。
另一個例子是衰老。果糖代謝與氧化壓力、粒線體功能障礙、細胞保護轉錄因子核因子红系2相關因子2(Nrf2)的損失以及表徵衰老過程中的sirtuins減少相關[50,189]。果糖還比葡萄糖更有效地誘導高級糖基化終產物的生成[190,191]。正如前面提到的,我們發現老年腎臟疾病在老年KHK-KO小鼠中不會發生[145]。
越來越多的證據表明,阿爾茨海默病可能與顱內果糖代謝有關[75]。阿爾茨海默病在那些飲食富含或產生果糖的食物(高糖、高鹽、高血糖和高加工紅肉飲食)或患有代謝綜合症和糖尿病特徵的人中增加。果糖的投予也可以在實驗室大鼠中誘導阿爾茨海默病的特徵(包括精神狀態下降和淀粉樣蛋白和τ蛋白沉積),並且早期阿爾茨海默病患者的大腦中果糖也升高(參考[75,88]中的綜述)。機制似乎是由於過量果糖的生物學效應,抑制了大腦的某些區域,以刺激覓食反應[75,88]。
一個關鍵發現是果糖途徑的作用在早期疾病狀態中幾乎不可避免地最強,因為隨著時間的推移,通常會出現纖維化、發炎或線粒體損失,從而使疾病過程持續存在(表1)。因此,最好的干預時機可能在疾病早期,以防止條件變得不可逆轉。然而,由於許多疾病代表代謝紊亂,其中線粒體功能受到抑制,因此一種理想的方法是通過運動或其他方式嘗試再生線粒體。在這方面,雖然低果糖和低鹽飲食都可以改善線粒體功能[192],但另一個出色的方法是通過運動[23]。
10 限制和挑戰
這個假說存在一些挑戰。首先,有爭議關於是否存在節儉基因假說,因為似乎大多數饑荒時間太短,無法提供足夠的自然選擇壓力,以致使生存基因出現,決定一個物種的命運(詳見[124,193]中的討論)。有人還主張,如果節儉基因存在,那麼今天的每個人都應該是肥胖的。然而,尿酸酶酶突變發生在持續數百萬年的季節性飢荒期間,並且該突變並未導致肥胖,而是防止了饑荒。我們的假說還表明,肥胖不會發生在每個人身上,因為它需要一種互動,其中一個人會變得對瘦素(來自果糖代謝)產生耐受性,然後攝入高能量食物(如脂肪)。
我們同意,在現代人類的時期,生存基因較不可能出現。儘管如此,冰河時代持續了5000至10000年,進一步減少了大型獵物的可用性,導致了饑荒和人口的收縮,這影響了薩滿主義和藝術的發展[194]。還有一次約4200年前的200年乾旱,導致多個王國的崩潰,並導致乳糖耐受基因的快速傳播,因為它具有生存優勢[195]。然而,現在更不太可能出現生存基因,並且增加的脂肪儲備更不太可能帶來好處,因此今天肥胖基因更可能通過隨機漂移傳播(隨機漂移基因假說[196]),特別是對於在文明社會中肥胖且久坐不動的人來說,被掠食的風險較小[196,197]。
也有一些證據表明,在過去十年中,飲用含糖飲料的攝入量已經減少,從而導致了添加糖的總攝入量減少[198]。然而,添加糖的攝入量仍然相當大,仍然占整體飲食攝入量的15%。加工食品仍然在70%以上的產品中含有過多的添加糖和鹽[199,200]。因此,暴露於含有或刺激果糖產生的食物的可能性仍然超過了在大多數人中誘導瘦素耐受所必需的閾值。
還有一些證據表明,脂肪在缺乏果糖的情況下可能單獨引起肥胖。我們注意到飽和脂肪(牛油)可以在缺乏果糖激酶的小鼠中引起輕度肥胖,並且這與輕度肝脂肪化相關[144]。由於其抑制脂肪分解並增加游離脂肪酸運送到肝臟的能力,飽和脂肪似乎能夠比其他脂肪更好地誘導肝脂肪化[201]。另一項研究發現,高動物脂肪(特別是牛油和奶酪)的飲食可能增加患糖尿病的風險[202]。然而,因紐特人在食用肉類和飽和脂肪的飲食中沒有肥胖和糖尿病的情況使我們認為這不是引起肥胖和胰島素抵抗的主要機制。事實上,飽和脂肪的攝入量在上個世紀下降,並且與肥胖,糖尿病和非酒精性脂肪肝的流行病學不相關[203],而非酒精性脂肪肝與含糖飲料的攝入量相關[204]。
另一個令人困惑的發現是,采用低碳水化合物的生酮飲食的受試者通常會因酮尿而出現高尿酸血症,這會干擾尿酸排泄。盡管高尿酸有時會導致痛風,但目前尚不清楚高尿酸是否還有其他有害影響。有人認為,高尿酸可能存在是為了刺激從氨基酸進行糖异生[48],但也有可能是由於酮血症的對抗效應[205]或者由於在低碳水化合物飲食環境中尿酸誘導的果糖生成不太可能。我們知道,在飢餓的情況下,以及很可能在嚴格的碳水化合物限制下,果糖不再刺激脂肪儲存,而是優先轉化為葡萄糖以滿足即時能量需求[52]。顯然,需要進一步的研究來探討采用生酮飲食的人群中高尿酸血症的作用。
最後,已知並非所有冬眠動物在觸發增加食物攝入的事件中攝取果糖,一些冬眠動物可能在同樣攝取相同飼料的情況下仍然會出現明顯的脂肪儲存增加的現象。目前尚不清楚這些動物的觸發因素是什麼,儘管可能與水分狀態的變化、食物不足、基因因素或其他未知機制有關[206-208]。我們確實知道,即使這些松鼠采用相同的飲食進入冬眠,它們的肝臟仍然會出現類似于果糖代謝的代謝變化,體重增加與肝臟中AMP脫氨酶和尿酸的激活有關,然後在冬眠期間氧化脂肪時刺激AMP激活的蛋白激酶[80]。顯然,需要進一步的研究。
11 可測試的預測
儘管對於果糖生存假說存在大量支持性證據,但也有可測試的預測。首先,有必要確認內源性果糖途徑在人群中的重要性,並確定對生物學結果的影響程度。一種可能的方法是開發可以干擾這一途徑的藥物,例如開發有效的KHK、AMP去氨酶-2或其他生存途徑中的酶的抑制劑。研究內源性果糖途徑在冬眠哺乳動物和長途徑遷徙的鳥類中的作用也將有助於進一步了解這一假說。第二步是更好地了解從早期較可逆轉的狀態轉變為較持久的狀態的機制。一種方法是識別可以記錄這一轉變的生物標記。例如,最有效地測試抗利尿激素途徑的重要性,可以通過在對代謝綜合症風險的受試者的初步檢查中評估血清copeptin,並進行一項研究,該研究可以通過水分和鹽和糖的限制或使用抗利尿激素1b受體拮抗劑來抑制它[93]。同樣,研究尿酸的作用可能在疾病早期的代謝特徵上最有可能顯示出益處[176,209]。
12 總結
肥胖及其相關的代謝性疾病對現代社會造成了毀滅性的健康影響。在這裡,我們將這些疾病與一個在自然界中發展出來的主要生存途徑相關聯,這個途徑旨在幫助動物為糧食短缺的時期做好準備。不幸的是,我們無意中採用了這些啟動這一途徑的食物作為我們日常飲食的一部分,再加上我們所擁有的節約基因,我們現在正在承受將這個生存途徑推向極限的後果。我們的成功悲劇比我們想象的還要大,因為新的研究表明,果糖途徑還可能增加我們患癌症、與懷孕有關的疾病和神經系統疾病的風險。我們建議繼續進行上述所述的研究,以更好地了解果糖代謝在健康和疾病中的作用。